Last active
March 23, 2018 10:08
-
-
Save trvinh/6208c21b409a776892a37333a686387b to your computer and use it in GitHub Desktop.
modified class2tree function & create a rooted taxonomy tree from a taxa list
This file contains bidirectional Unicode text that may be interpreted or compiled differently than what appears below. To review, open the file in an editor that reveals hidden Unicode characters.
Learn more about bidirectional Unicode characters
| #' Convert a list of classifications to a tree. | |
| #' | |
| #' This function converts a list of hierarchies for individual species into | |
| #' a single species by taxonomic level matrix, then calculates a distance | |
| #' matrix based on taxonomy alone, and outputs either a phylo or dist object. | |
| #' See details for more information. | |
| #' | |
| #' @export | |
| #' @param input List of classification data.frame's from the function | |
| #' \code{\link{classification}} | |
| #' @param varstep Vary step lengths between successive levels relative to | |
| #' proportional loss of the number of distinct classes. | |
| #' @param check If TRUE, remove all redundant levels which are different for | |
| #' all rows or constant for all rows and regard each row as a different basal | |
| #' taxon (species). If FALSE all levels are retained and basal taxa (species) | |
| #' also must be coded as variables (columns). You will get a warning if | |
| #' species are not coded, but you can ignore this if that was your intention. | |
| #' @param ... Further arguments passed on to hclust. | |
| #' @param x Input object to print or plot - output from class2tree function. | |
| #' @return An object of class "classtree" with slots: | |
| #' \itemize{ | |
| #' \item phylo - The resulting object, a phylo object | |
| #' \item classification - The classification data.frame, with taxa as rows, | |
| #' and different classification levels as columns | |
| #' \item distmat - Distance matrix | |
| #' \item names - The names of the tips of the phylogeny | |
| #' } | |
| #' | |
| #' Note that when you execute the resulting object, you only get the phylo | |
| #' object. You can get to the other 3 slots by calling them directly, like | |
| #' output$names, etc. | |
| #' @details See \code{\link[vegan]{taxa2dist}}. Thanks to Jari Oksanen for | |
| #' making the taxa2dist function and pointing it out, and Clarke & Warwick | |
| #' (1998, 2001), which taxa2dist was based on. | |
| #' @examples \dontrun{ | |
| #' spnames <- c('Quercus robur', 'Iris oratoria', 'Arachis paraguariensis', | |
| #' 'Helianthus annuus','Madia elegans','Lupinus albicaulis', | |
| #' 'Pinus lambertiana') | |
| #' out <- classification(spnames, db='itis') | |
| #' tr <- class2tree(out) | |
| #' plot(tr) | |
| #' | |
| #' spnames <- c('Klattia flava', 'Trollius sibiricus', 'Arachis paraguariensis', | |
| #' 'Tanacetum boreale', 'Gentiana yakushimensis','Sesamum schinzianum', | |
| #' 'Pilea verrucosa','Tibouchina striphnocalyx','Lycium dasystemum', | |
| #' 'Berkheya echinacea','Androcymbium villosum', | |
| #' 'Helianthus annuus','Madia elegans','Lupinus albicaulis', | |
| #' 'Pinus lambertiana') | |
| #' out <- classification(spnames, db='ncbi') | |
| #' tr <- class2tree(out) | |
| #' plot(tr) | |
| #' } | |
| class2tree <- function(input, varstep = TRUE, check = TRUE, ...) { | |
| if (any(is.na(input))) { | |
| message('Removed species without classification.') | |
| input <- input[!is.na(input)] | |
| } | |
| # Check that there is more than 2 taxon | |
| if (length(input) < 3) | |
| stop("Your input list of classifications must be 3 or longer.") | |
| if (length(unique(names(input))) < length(names(input))) | |
| stop("Input list of classifications contains duplicates") | |
| dat <- rbind.fill(lapply(input, class2tree_helper)) | |
| # Get rank and ID list | |
| rankList <- rbind.fill(lapply(input, get_rank)) | |
| nameList <- rbind.fill(lapply(input, get_name)) | |
| # Create taxonomy matrix | |
| df <- taxonomy_table_creator(nameList,rankList) | |
| if (!inherits(df, "data.frame")) { | |
| stop("no taxon ranks in common - try different inputs") | |
| } | |
| row.names(df) <- df[,1] | |
| df <- df[,-1] | |
| # calculate distance matrix | |
| taxdis <- tryCatch(taxa2dist(df, varstep = varstep, check = check), | |
| error = function(e) e) | |
| tdf = t(df) | |
| for (i in 1:ncol(tdf)){ | |
| tdf[,i][duplicated(tdf[,i])] <- NA | |
| } | |
| # check for incorrect dimensions error | |
| if (is(taxdis, 'simpleError')) | |
| stop("Try check=FALSE, but see docs for taxa2dist function in the vegan package for details.") | |
| out <- as.phylo.hclust(hclust(taxdis, ...)) | |
| res <- list(phylo = out, classification = as.data.frame(t(tdf)), distmat = taxdis, | |
| names = names(input)) | |
| class(res) <- 'classtree' | |
| return( res ) | |
| } | |
| class2tree_helper <- function(x){ | |
| df <- x[-nrow(x), 'name'] | |
| names(df) <- x[-nrow(x), 'rank'] | |
| df <- data.frame(t(data.frame(df)), stringsAsFactors = FALSE) | |
| data.frame(tip = x[nrow(x), "name"], df, stringsAsFactors = FALSE) | |
| } | |
| #' @method plot classtree | |
| #' @export | |
| #' @rdname class2tree | |
| plot.classtree <- function(x, ...) { | |
| if (!is(x$phylo, "phylo")) | |
| stop("Input object must have a slot in 'phylo' of class 'phylo'") | |
| plot(x$phylo, ...) | |
| } | |
| #' @method print classtree | |
| #' @export | |
| #' @rdname class2tree | |
| print.classtree <- function(x, ...) { | |
| if (!is(x$phylo, "phylo")) | |
| stop("Input object must have a slot in 'phylo' of class 'phylo'") | |
| print(x$phylo) | |
| } | |
| # Function from the vegan package | |
| # CRAN: http://cran.rstudio.com/web/packages/vegan/ | |
| # License: GPL-2 | |
| # Maintainer: Jari Oksanen <jari.oksanen at oulu.fi> | |
| taxa2dist <- function(x, varstep = FALSE, check = TRUE, labels) { | |
| rich <- apply(x, 2, function(taxa) length(unique(taxa))) | |
| S <- nrow(x) | |
| if (check) { | |
| keep <- rich < S & rich > 1 | |
| rich <- rich[keep] | |
| x <- x[, keep] | |
| } | |
| i <- rev(order(rich)) | |
| x <- x[, i] | |
| rich <- rich[i] | |
| if (varstep) { | |
| add <- -diff(c(nrow(x), rich, 1)) | |
| add <- add/c(S, rich) | |
| add <- add/sum(add) * 100 | |
| } | |
| else { | |
| add <- rep(100/(ncol(x) + check), ncol(x) + check) | |
| } | |
| if (!is.null(names(add))) | |
| names(add) <- c("Base", names(add)[-length(add)]) | |
| if (!check) | |
| add <- c(0, add) | |
| out <- matrix(add[1], nrow(x), nrow(x)) | |
| for (i in 1:ncol(x)) { | |
| out <- out + add[i + 1] * outer(x[, i], x[, i], "!=") | |
| } | |
| out <- as.dist(out) | |
| attr(out, "method") <- "taxa2dist" | |
| attr(out, "steps") <- add | |
| if (missing(labels)) { | |
| attr(out, "Labels") <- rownames(x) | |
| } | |
| else { | |
| if (length(labels) != nrow(x)) | |
| warning("Labels are wrong: needed ", nrow(x), " got ", | |
| length(labels)) | |
| attr(out, "Labels") <- as.character(labels) | |
| } | |
| if (!check && any(out <= 0)) | |
| warning("you used 'check=FALSE' and some distances are zero -- was this intended?") | |
| out | |
| } | |
| ############################################################################### | |
| #################### GET LIST OF ALL TAXONOMY RANK AND IDs #################### | |
| ############################################################################### | |
| get_rank <- function(x){ | |
| rankDf <- x[, 'rank'] | |
| names(rankDf) <- x[, 'rank'] | |
| idDf <- x[, 'id'] | |
| joinedDf <- cbind(data.frame(rankDf,stringsAsFactors=FALSE), | |
| data.frame(idDf,stringsAsFactors=FALSE)) | |
| joinedDf <- within(joinedDf, | |
| rankDf[rankDf=='no rank'] <- | |
| paste0("norank_",idDf[rankDf=='no rank'])) | |
| df <- data.frame(t(data.frame(rev(joinedDf$rankDf))), | |
| stringsAsFactors = FALSE) | |
| outDf <- data.frame(tip = x[nrow(x), "name"], df, stringsAsFactors = FALSE) | |
| return(outDf) | |
| } | |
| get_name <- function(x){ | |
| rankDf <- x[, 'rank'] | |
| names(rankDf) <- x[, 'rank'] | |
| nameDf <- x[, 'name'] | |
| idDf <- x[, 'id'] | |
| joinedDf <- cbind(data.frame(rankDf,stringsAsFactors=FALSE), | |
| data.frame(nameDf,stringsAsFactors=FALSE)) | |
| joinedDf <- within(joinedDf, | |
| rankDf[rankDf=='no rank'] <- | |
| paste0("norank_",idDf[rankDf=='no rank'])) | |
| joinedDf$name <- paste0(joinedDf$nameDf,"#",joinedDf$rankDf) | |
| df <- data.frame(t(data.frame(rev(joinedDf$name))), stringsAsFactors = FALSE) | |
| outDf <- data.frame(tip = x[nrow(x), "name"], df, stringsAsFactors = FALSE) | |
| return(outDf) | |
| } | |
| ############################################################################### | |
| #################### INDEXING ALL AVAILABLE RANKS (INCLUDING NORANK) ########## | |
| ############################################################################### | |
| rank_indexing <- function(rankList){ | |
| ##### input is a dataframe, where each row is a rank list of a taxon | |
| ### get all available ranks from input rankList | |
| uList <- unlist(rankList) | |
| # remove unique rank by replacing with NA | |
| # (unique rank is uninformative for sorting taxa) | |
| uList[!duplicated(uList)] <- NA | |
| # get final list of available ranks (remove NA items) | |
| allInputRank <- as.character(unique(uList)) | |
| allInputRank <- allInputRank[!is.na(allInputRank)] | |
| ### initial index for main ranks | |
| mainRank <- c("strain","forma","subspecies","varietas","species", | |
| "speciessubgroup","speciesgroup","subgenus","genus","subtribe", | |
| "tribe","subfamily","family","superfamily","parvorder", | |
| "infraorder","suborder","order","superorder","infraclass", | |
| "subclass","class","superclass","subphylum","phylum", | |
| "superphylum","subkingdom","kingdom","superkingdom") | |
| rank2Index <- new.env() | |
| for(i in 1:length(mainRank)){ | |
| rank2Index[[mainRank[i]]] = i | |
| } | |
| ### the magic happens here | |
| for(k in 1:nrow(rankList)){ | |
| ### get subset of rank list for current taxon which | |
| ### contains only ranks existing in allInputRank | |
| subList <- rankList[k,][!is.na(rankList[k,])] | |
| filter <- sapply(subList, function(x) x %in% allInputRank) | |
| subList <- subList[filter] | |
| ### now go to each rank and check... | |
| for(i in 1:length(subList)){ | |
| ## if it has no index (probably only for norank), then... | |
| if(is.null(rank2Index[[subList[i]]])){ | |
| ## set index for this rank = the [available] index of previous rank + 1 | |
| for(j in 1:length(subList)){ | |
| if(!is.null(rank2Index[[subList[i-j]]])){ | |
| rank2Index[[subList[i]]] = rank2Index[[subList[i-j]]] + 1 | |
| break | |
| } else { | |
| j = j-1 | |
| } | |
| } | |
| } | |
| ## else, check if the current index is smaller than | |
| ## the index of previous rank, | |
| else { | |
| if(i>1){ | |
| preRank <- subList[i-1] | |
| ## if so, increase index of this current rank | |
| ## by (index of previous rank + 1) | |
| if(rank2Index[[subList[i]]] <= rank2Index[[preRank]]){ | |
| rank2Index[[subList[i]]] = rank2Index[[preRank]] + 1 | |
| } | |
| } | |
| } | |
| } | |
| } | |
| ### output a list of indexed ranks | |
| index2RankDf <- data.frame("index"=character(), | |
| "rank"=character(), | |
| stringsAsFactors=FALSE) | |
| for(i in 1:length(allInputRank)){ | |
| index2RankDf[i,] = c(rank2Index[[allInputRank[i]]],allInputRank[i]) | |
| } | |
| index2RankDf$index <- as.numeric(index2RankDf$index) | |
| index2RankDf <- index2RankDf[with(index2RankDf, order(index2RankDf$index)),] | |
| return(index2RankDf) | |
| } | |
| ############################################################################### | |
| #################### ARRANGE RANK IDs INTO SORTED RANK LIST (index2RankDf) #### | |
| ############################################################################### | |
| taxonomy_table_creator <- function(nameList,rankList){ | |
| colnames(nameList)[1] <- "tip" | |
| ### get indexed rank list | |
| index2RankDf <- rank_indexing(rankList) | |
| ### get ordered rank list | |
| orderedRank <- factor(index2RankDf$rank, levels = index2RankDf$rank) | |
| ### create a dataframe containing ordered ranks | |
| full_rank_name_df <- data.frame("rank"=matrix(unlist(orderedRank), | |
| nrow=length(orderedRank), | |
| byrow=TRUE), | |
| stringsAsFactors=FALSE) | |
| full_rank_name_df$index <- as.numeric(rownames(full_rank_name_df)) | |
| for(i in 1:nrow(nameList)){ | |
| ### get list of all IDs for this taxon | |
| taxonDf <- data.frame(nameList[i,]) | |
| taxonName <- unlist(strsplit(as.character(nameList[i,]$tip), | |
| "#", | |
| fixed = TRUE)) | |
| ### convert into long format | |
| mTaxonDf <- suppressWarnings(melt(taxonDf,id = "tip")) | |
| ### get rank names and corresponding IDs | |
| splitCol <- data.frame(do.call('rbind', | |
| strsplit(as.character(mTaxonDf$value), | |
| '#', | |
| fixed=TRUE))) | |
| mTaxonDf <- cbind(mTaxonDf,splitCol) | |
| ### remove NA cases | |
| mTaxonDf <- mTaxonDf[complete.cases(mTaxonDf),] | |
| ### subselect mTaxonDf to keep only 2 column rank id and rank name | |
| mTaxonDf <- mTaxonDf[,c("X1","X2")] | |
| if(mTaxonDf$X2[1] != index2RankDf$rank[1]){ | |
| mTaxonDf <- rbind(data.frame("X1"=mTaxonDf$X1[1], | |
| "X2"=index2RankDf$rank[1]), | |
| mTaxonDf) | |
| } | |
| ### rename columns | |
| colnames(mTaxonDf) <- c(taxonName[1],"rank") | |
| ### merge with index2RankDf (Df contains all available ranks of input data) | |
| full_rank_name_df <- merge(full_rank_name_df,mTaxonDf, by=c("rank"), all.x = T) | |
| ### reorder ranks | |
| full_rank_name_df <- full_rank_name_df[order(full_rank_name_df$index),] | |
| ### replace NA id by id of previous rank | |
| full_rank_name_df <- na.locf(full_rank_name_df) | |
| } | |
| ### remove index column | |
| full_rank_name_df <- subset(full_rank_name_df, select = -c(index)) | |
| ### transpose into wide format | |
| t_full_rank_name_df <- transpose(full_rank_name_df) | |
| ### set first row to column names | |
| colnames(t_full_rank_name_df) = as.character(unlist(t_full_rank_name_df[1,])) | |
| t_full_rank_name_df <- t_full_rank_name_df[-1,] | |
| ### replace NA values in the dataframe t_full_rank_name_df | |
| if(nrow(t_full_rank_name_df[is.na(t_full_rank_name_df),]) > 0){ | |
| t_full_rank_name_dfTMP <- t_full_rank_name_df[complete.cases(t_full_rank_name_df),] | |
| t_full_rank_name_dfEdit <- t_full_rank_name_df[is.na(t_full_rank_name_df),] | |
| for(i in 1:nrow(t_full_rank_name_dfEdit)){ | |
| for(j in 1:(ncol(t_full_rank_name_df)-1)){ | |
| if(is.na(t_full_rank_name_dfEdit[i,j])){ | |
| t_full_rank_name_dfEdit[i,j] <- t_full_rank_name_dfEdit[i,j+1] | |
| } | |
| } | |
| if(is.na(t_full_rank_name_dfEdit[i,ncol(t_full_rank_name_df)])){ | |
| t_full_rank_name_dfEdit[i,ncol(t_full_rank_name_df)] <- | |
| t_full_rank_name_dfEdit[i,ncol(t_full_rank_name_df)-1] | |
| } | |
| } | |
| t_full_rank_name_df <- rbind(t_full_rank_name_dfEdit,t_full_rank_name_dfTMP) | |
| } | |
| ### add fullName column into t_full_rank_name_df | |
| fullName <- colnames(full_rank_name_df)[-c(1)] | |
| full_rank_name_df <- cbind(fullName,t_full_rank_name_df) | |
| ### return | |
| return(full_rank_name_df) | |
| } |
Output tree
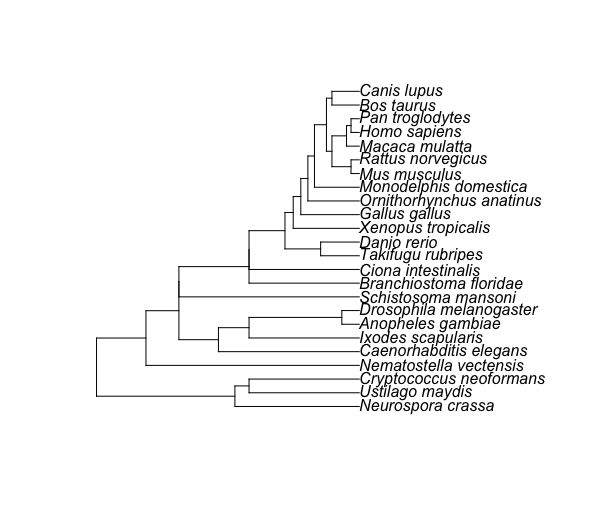
this modified code already merged to taxize 🥇
rooting the tree using a reference species
# get rank and name lists
rankList <- rbind.fill(lapply(outTaxize, get_rank))
nameList <- rbind.fill(lapply(outTaxize, get_name))
# create DF for calculating distance matrix
df <- taxonomy_table_creator(nameList, rankList
row.names(df) <- df[,1]
df <- df[,-1]
# calculate distance matrix
taxDis <- tryCatch(taxa2dist(df), error = function(e) e)
# generate phylo tree object
tree <- as.phylo(hclust(taxDis))
# root the tree
tree <- root(tree, outgroup = "Homo sapiens", resolve.root = TRUE)
#plot(tree, align.tip.label = TRUE, use.edge.length = TRUE)
# export tree in newick format
write.tree(tree, file = "outputTree.nw", append = FALSE, digits = 10, tree.names = FALSE)
# get ordered taxon list and print to file
is_tip <- tree$edge[,2] <= length(tree$tip.label)
ordered_tips <- tree$edge[is_tip, 2]
sortedTaxonList <- rev(tree$tip.label[ordered_tips])
write.table(sortedTaxonList, file ="sortedTaxonList.txt", col.names = F, row.names = F, quote = F, sep="\t")
Results:
- Sorted taxon list
> print(taxonList)
[1] "Homo sapiens" "Pan troglodytes" "Macaca mulatta" "Canis lupus"
[5] "Bos taurus" "Rattus norvegicus" "Mus musculus" "Monodelphis domestica"
[9] "Ornithorhynchus anatinus" "Gallus gallus" "Xenopus tropicalis" "Danio rerio"
[13] "Takifugu rubripes" "Branchiostoma floridae" "Ciona intestinalis" "Schistosoma mansoni"
[17] "Drosophila melanogaster" "Anopheles gambiae" "Ixodes scapularis" "Caenorhabditis elegans"
[21] "Nematostella vectensis" "Cryptococcus neoformans" "Ustilago maydis" "Neurospora crassa"
- and the taxonomy tree (note: it looks different from the tree of
taxize)
> plot(tree)

Sign up for free
to join this conversation on GitHub.
Already have an account?
Sign in to comment
Usage example: