- Perform single-sample germline variant calling with GATK HaplotypeCaller and the GATK GVCF workflow on exome data
- Perform joint genotype calling on exome data, including additional exomes from 1000 Genomes Project
- Manipulate and Filter VCFs to remove artifacts and identify variants of interest
In this module we will use the GATK HaplotypeCaller to call germline variants from "normal" bams. For a more in-depth look, see this excellent GATK tutorial, provided by the Broad Institute.
Note: We are using GATK v4.2.1.0 for this tutorial.
Grab some bam files from the HCC1395 "normal" cell line and save them:
cd /workspace
wget https://storage.googleapis.com/bfx_workshop_tmp/align.tar
tar -xvf align.tar
Make a folder called germline and cd into it
mkdir germline && cd germline
First, we will run GATK HaplotypeCaller to call germline SNPs and indels. Whenever HaplotypeCaller finds signs of variation it performs a local de novo re-assembly of reads. This improves the accuracy of variant calling, especially in challenging regions. We'll also include an option to generate bam output from HaplotypeCaller so that local reassembly/realignment around called variants can be visualized.
We'll run GATK HaplotypeCaller with the following options:
--java-options '-Xmx12g'tells GATK to use 12GB of memoryHaplotypeCallerspecifies the GATK command to run-Rspecifies the path to the reference genome-Ispecifies the path to the input bam file for which to call variants-Ospecifies the path to the output vcf file to write variants to--bam-outputspecifies the path to an optional bam file which will store local realignments around variants that HaplotypeCaller used to make calls-Lis used to limit calling to specific regions (e.g. just chr17). It cat unwieldy when this list of regions gets long, so we often use an environment variable to handle this. In this case, we've pre-loaded one called$GATK_REGIONS. To verify it's contents, you can runecho $GATK_VARIANTS
All right, let's run this tool:
#Create working dir for germline results if not already created
mkdir -p /workspace/germline
cd /workspace/germline
#Call variants for exome data
gatk --java-options '-Xmx12g' HaplotypeCaller -R /workspace/inputs/references/genome/ref_genome.fa -I /workspace/align/Exome_Norm_sorted_mrkdup_bqsr.bam -O /workspace/germline/Exome_Norm_HC_calls.vcf --bam-output /workspace/germline/Exome_Norm_HC_out.bam $GATK_REGIONSCalling variants for the WGS data is going to take a while. Let's run it in the background and pipe the results to files so that we can keep working at the terminal:
gatk --java-options '-Xmx7g' HaplotypeCaller -R /workspace/inputs/references/genome/ref_genome.fa -I /workspace/align/WGS_Norm_merged_sorted_mrkdup_bqsr.bam -O /workspace/germline/WGS_Norm_HC_calls.vcf --bam-output /workspace/germline/WGS_Norm_HC_out.bam $GATK_REGIONS 2>hc_wgs.err >hc_wgs.out &
Let's examine and compare the alignments made by HaplotypeCaller for local re-alignment around variants to those produced originally by BWA-MEM for the normal (germline) exome data. First start a new IGV session and load the following bam files:
- Exome_Norm_sorted_mrkdup_bqsr.bam
- Exome_Norm_HC_out.bam
We identified an illustrative deletion by looking through the germline variant calls that we just created in Exome_Norm_HC_calls.vcf. Once you have loaded the above bam files and performed any desired set up (e.g., naming your tracks) navigate to the following coordinates: chr17:76286974-76287203.
You should see something like the following:
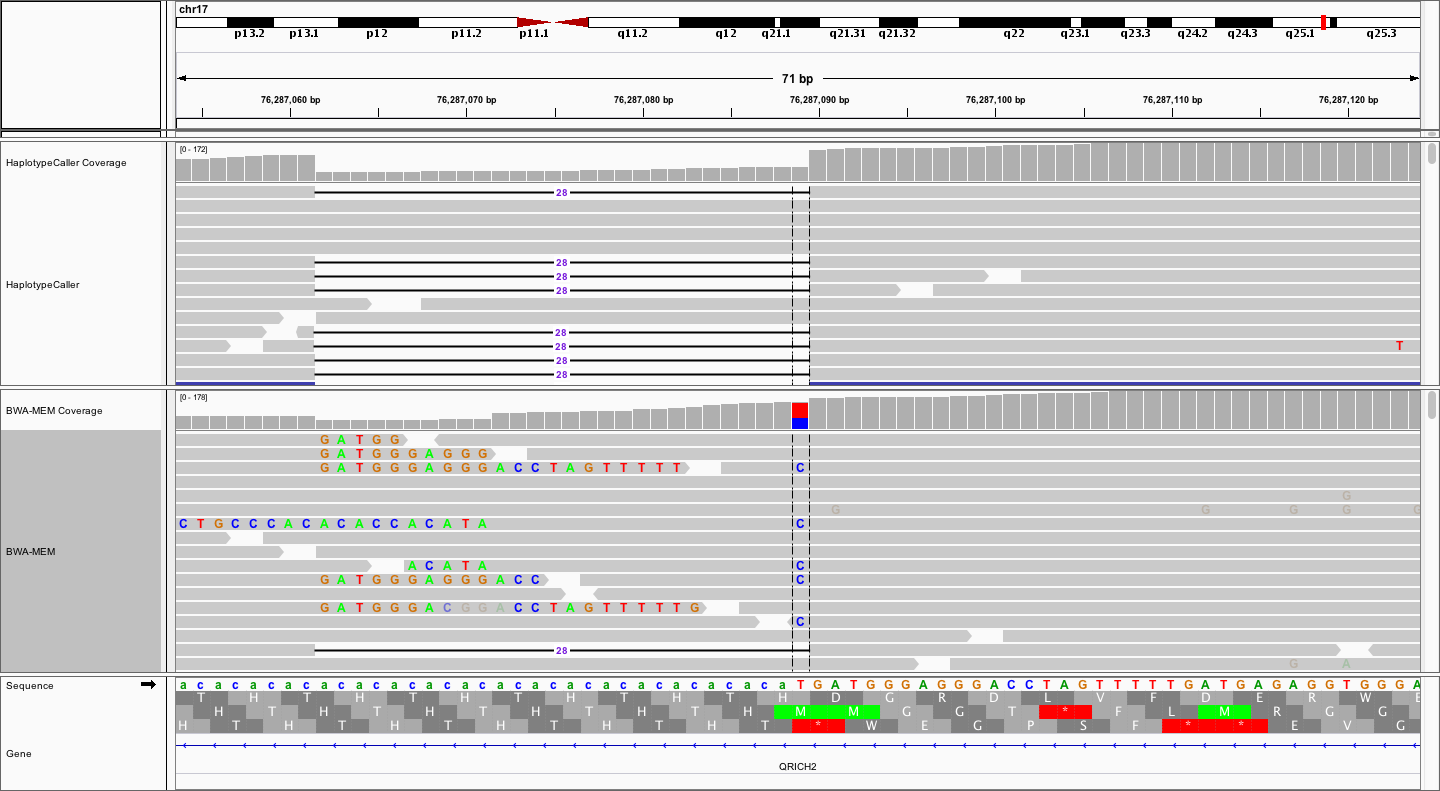
Notice that the local realignment (top track) has produced a very clear call of a 28bp deletion. In contrast, the bwa-mem alignment, while also supporting the 28bp deletion with some reads, is characterized by a bunch of messy soft-clipping and a probably false positive SNV at the end of CA repeat. The contrast is even more striking when we collapse all reads to a squished view.
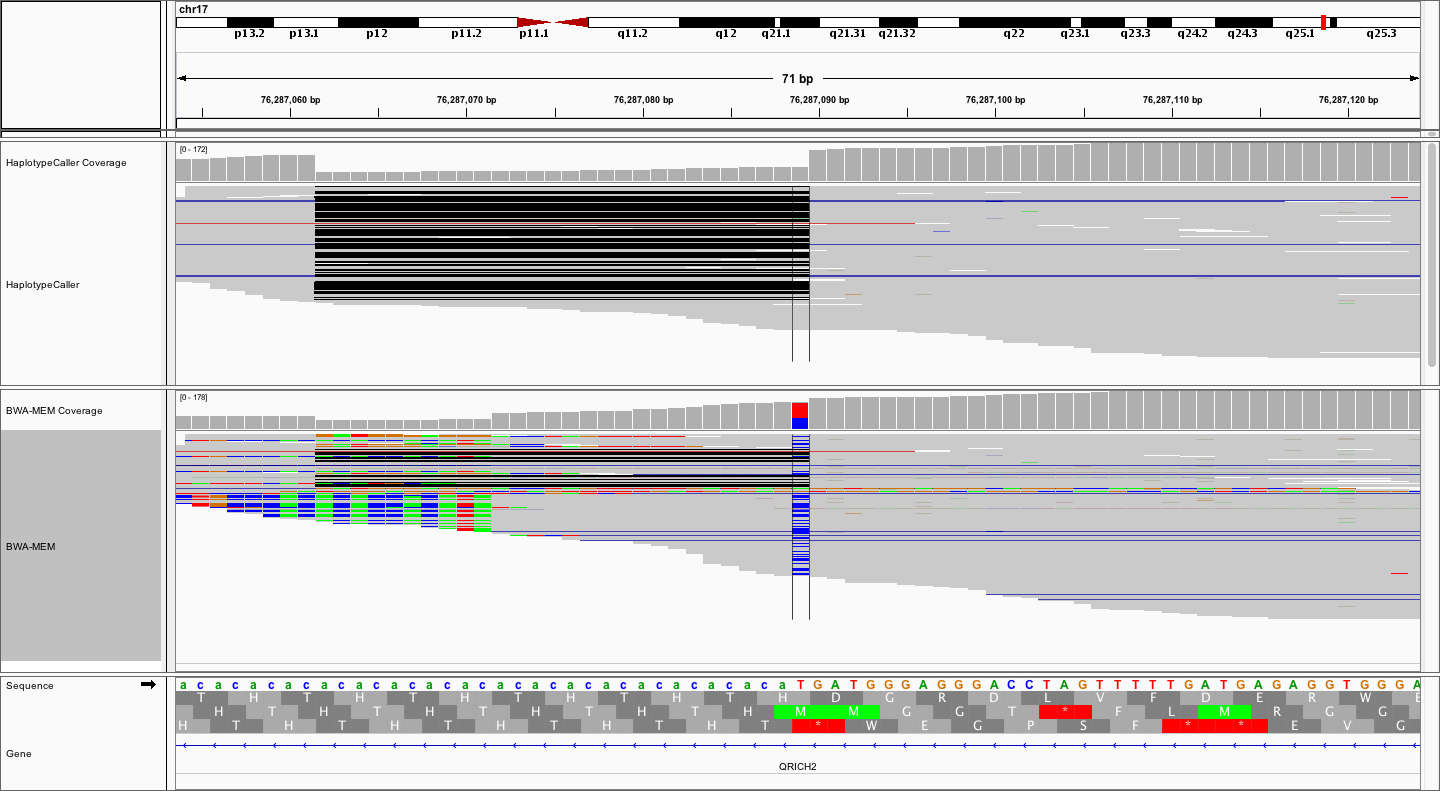
Notice how much "cleaner" the alignments are in the top (locally realigned) track. Unfortunately, many variant callers (without local realignment) would likely call the T->C variant, requiring subsequent filtering and/or manual review to eliminate this artifact. Finally, let's zoom out a little more to get the big picture.
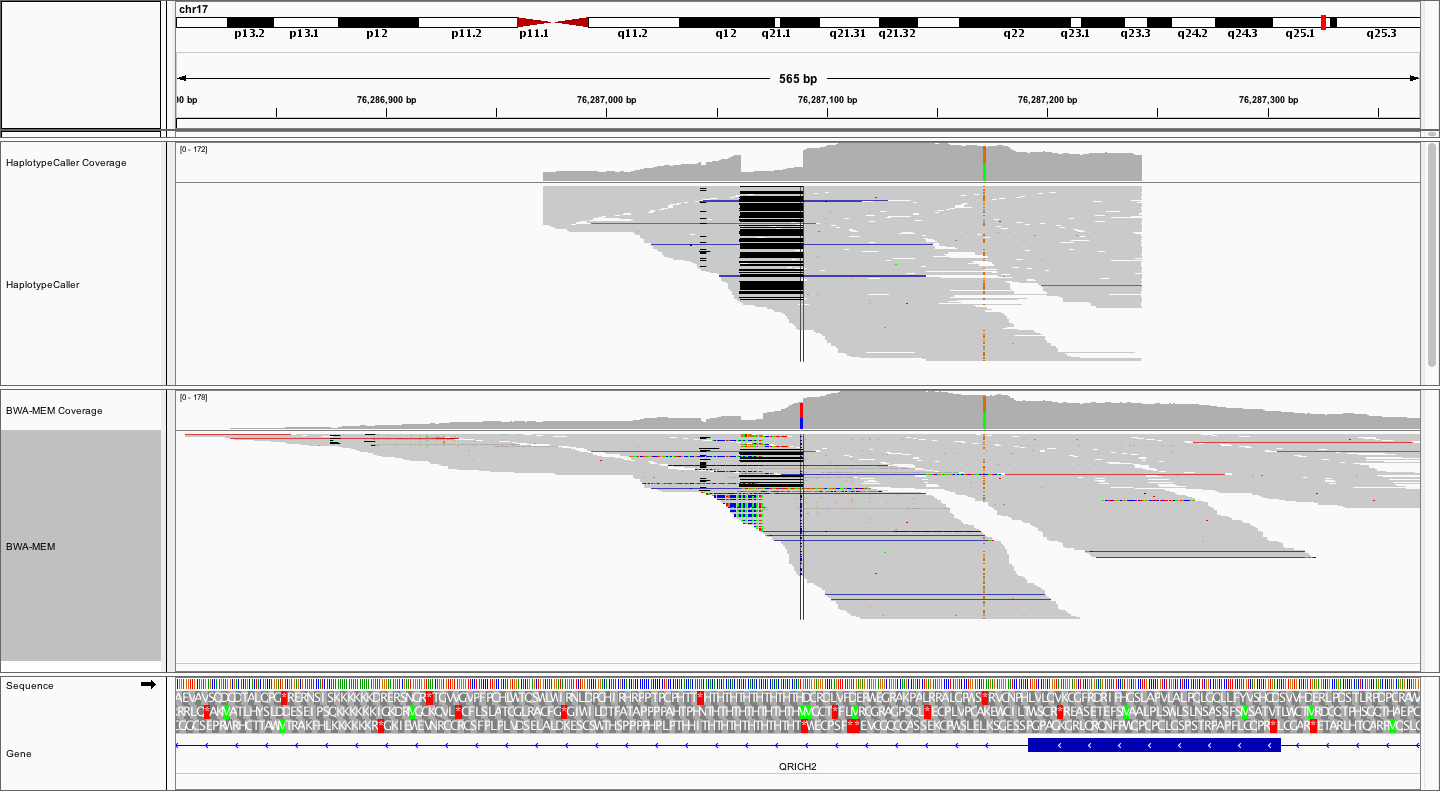
What differences do you notice between the local realignment and bwa-mem alignment from this perspective, apart from the already observed more clean 28bp deletion?
Answer
In the local realignment, the alignments stop abruptly, obviously within some pre-determined window. On the other hand the bwa-mem alignments spread out further, and are naturally distributed around the targeted region (exon). Remember that the local alignment is *local*, not complete. This means that the bam files from HaplotypeCaller represent only a subset of alignments, centered around potential variation. These BAM files are therefore not a replacement for the complete bwa-mem BAMs.
An alternate (and GATK recommended) method is to use the GVCF workflow. In this mode, HaplotypeCaller runs per-sample to generate an intermediate GVCF. These intermediate GVCFs can then be used with the GenotypeGVCF command for joint genotyping of multiple samples in a very efficient way. Joint genotyping has several advantages:
- In joint genotyping, variants are analyzed across all samples simultaneously. This ensures that if any sample in the study population has variation at a specific site then the genotype for all samples will be determined at that site.
- It allows more sensitive detection of genotype calls at sites where one sample might have lower coverage but other samples have a confident variant at that position.
- Joint calling is also necessary to produce data needed for VQSR filtering (see [next module]({{ site.baseurl }}{% link _posts/0004-02-02-Germline_SnvIndel_FilteringAnnotationReview.md %})).
While joint genotyping can be performed directly with HaplotypeCaller (by specifying multiple -I paramters) it is less efficient, and less scalable. Each time a new sample is added the entire (progressively slower) variant calling analysis must be repeated. With the GVCF workflow, new samples can be run individually and then more quickly joint genotyped using the GenotypeGVCFs command.
GATK HaplotypeCaller is run with all of the same options as above, except for one addition:
- -ERC GVCF specifies to use the GCVF workflow, producing a GVCF instead of VCF
#Call variants in GVCF mode for exome data
gatk --java-options '-Xmx7g' HaplotypeCaller -ERC GVCF -R /workspace/inputs/references/genome/ref_genome.fa -I /workspace/align/Exome_Norm_sorted_mrkdup_bqsr.bam -O /workspace/germline/Exome_Norm_HC_calls.g.vcf --bam-output /workspace/germline/Exome_Norm_HC_GVCF_out.bam $GATK_REGIONSAs described above, there are several advantages to joint genotype calling. The additional samples and their variants improve the overall accuracy of variant calling and subsequent filtering steps. However, in this case we only have one "normal" sample, our HCC1395BL cell line. Therefore, we will make use of a set of public exome sequencing data to illustrate multi-sample joint calling. The 1000 Genome Project is a large international effort to create a set of publicly available sequence data for a large cohort of ethnically diverse individuals. Recall that the cell line being used in this tutorial (HCC1395) was derived from a 43 year old caucasian female (by a research group in Dallas Texas). Therefore, for a "matched" set of reference alignments we might limit to those of European descent.
As you go looking for this data, keep in mind the FAIR principles that we talked about earlier in the course (Findable, Accessible, Interoperable, Reproducible)
-
Hop onto the 1000 Genomes Samples page and scroll through the list of samples available.
-
In a real study, you'd spend a bunch of time thinking about what the proper samples to use were, to make sure that you're finding disease-related variants and not just population markers. In this case, we'll just assume that a few samples from the British in England and Scotland population will be a reasonable match.
-
Filter by population -> "British in England and Scotland"
-
Filter by technology -> Exome
-
Filter by data collection -> 1000 genomes on GRCh38
-
Download the list to get sample details (e.g., save as igsr_samples_GBR.tsv)
-
Open up this file and take a look. Does this have enough data to get at the variant calls? (Findable!)
- Click on "Data Collections" at the top
- Choose "1000 Genomes on GRCh38"
- In the Populations box, choose "British in England and Scotland"
- Scroll down to the bottom and filter the file list to Exome and Alignments
- 'Download the list' (e.g., save as igsr_GBR_GRCh38_exome_alignment.tsv).
With this list, we could download the already aligned exome data for a bunch of 1KGP individuals in cram format. (Accessible!) How can we tell whether these data files were generated in a way compatible with the data we've generated? Spend a minute or two looking before you check the answer below.
Answer
This takes a little digging. On the 1000 genomes site, go to the top, then click through to Help -> FAQ -> :What methods were used for generating alignments?". That directs you back to the [data collection page](https://www.internationalgenome.org/data-portal/data-collection), and for this "1000 Genomes on GRCh38" study, to the [latest 2019 publication here](https://wellcomeopenresearch.org/articles/4-50).
That was quite a journey, but if you scroll down, you'll notice that they do have excellent documentation of the tools, input files, and parameters used. (Reproducible!)
With this info, we can determine that these cram files were created in a generally compatible way with the anlysis done so far in this tutorial. (Interoperable!)
We still need to consider that there could be batch effects related to potentially different sample preparation, library construction, exome capture reagent and protocol, sequencing pipeline, etc. These are complex topics that need to built into the statistical models, and are a bit beyond the scope of what we're going to talk about in this course!
For expediency, in preparation for this course, we downloaded exome alignments for 5 females, of GBR descent. We limited that data to just chr6 and/or chr17 and then performed germline variant calling using the GATK HaplotypeCaller GVCF workflow commands, as above. The details for this can be found on the Developer's Notes page.
Download pre-computed GVCFs and their index files for five 1KGP exomes that we will use for joint genotyping.
cd /workspace/germline/
wget http://genomedata.org/pmbio-workshop/references/1KGP/gvcfs/chr17/HG00099_HC_calls.g.vcf
wget http://genomedata.org/pmbio-workshop/references/1KGP/gvcfs/chr17/HG00099_HC_calls.g.vcf.idx
wget http://genomedata.org/pmbio-workshop/references/1KGP/gvcfs/chr17/HG00102_HC_calls.g.vcf
wget http://genomedata.org/pmbio-workshop/references/1KGP/gvcfs/chr17/HG00102_HC_calls.g.vcf.idx
wget http://genomedata.org/pmbio-workshop/references/1KGP/gvcfs/chr17/HG00104_HC_calls.g.vcf
wget http://genomedata.org/pmbio-workshop/references/1KGP/gvcfs/chr17/HG00104_HC_calls.g.vcf.idx
wget http://genomedata.org/pmbio-workshop/references/1KGP/gvcfs/chr17/HG00150_HC_calls.g.vcf
wget http://genomedata.org/pmbio-workshop/references/1KGP/gvcfs/chr17/HG00150_HC_calls.g.vcf.idx
wget http://genomedata.org/pmbio-workshop/references/1KGP/gvcfs/chr17/HG00158_HC_calls.g.vcf
wget http://genomedata.org/pmbio-workshop/references/1KGP/gvcfs/chr17/HG00158_HC_calls.g.vcf.idxCreate a joint genotype GVCF, combining HCC1395 Exome normal together with a set of 1KGP exomes, for later use in VQSR filtering.
First, combine the g.vcf files together into a single combined g.vcf with the CombineGVCFs command.
GATK CombineGVCFs is run with the following options:
--java-options '-Xmx8g'tells GATK to use 12GB of memoryCombineGVCFsspecifies the GATK command to run-Rspecifies the path to the reference genome-V[multiple] specifies the path to each of the input g.vcf files-Ospecifies the path to the output combined g.vcf file$GATK_REGIONSprovides our-Lparams to limit the calling to specific regions
gatk --java-options '-Xmx8g' CombineGVCFs -R /workspace/inputs/references/genome/ref_genome.fa -V /workspace/germline/Exome_Norm_HC_calls.g.vcf -V /workspace/germline/HG00099_HC_calls.g.vcf -V /workspace/germline/HG00102_HC_calls.g.vcf -V /workspace/germline/HG00104_HC_calls.g.vcf -V /workspace/germline/HG00150_HC_calls.g.vcf -V /workspace/germline/HG00158_HC_calls.g.vcf -O /workspace/germline/Exome_Norm_1KGP_HC_calls_combined.g.vcf $GATK_REGIONSFinally, perfom joint genotyping with the GenotypeGVCFs command. This command is run with the following options:
- --java-options '-Xmx12g' tells GATK to use 12GB of memory
- GenotypeGVCFs specifies the GATK command to run
- -R specifies the path to the reference genome
- -V specifies the path to combined g.vcf files
- -O specifies the path to the output the joint genotyped vcf file
- $GATK_REGIONS is an environment variable that we defined earlier to limit calling to specific regions (e.g., just chr17)
gatk --java-options '-Xmx12g' GenotypeGVCFs -R /workspace/inputs/references/genome/ref_genome.fa -V /workspace/germline/Exome_Norm_1KGP_HC_calls_combined.g.vcf -O /workspace/germline/Exome_GGVCFs_jointcalls.vcf $GATK_REGIONSLook at this joint genotyping VCF (Exome_GGVCFs_jointcalls.vcf) and compare it to the "standard" VCF you created above (Exome_Norm_HC_calls.vcf). What are the key differences?
Let's clean up this directory a bit to make our life easier:
mkdir 1kgenomes
mv HG0* 1kgenomes
Check in on the WGS data - look at the log/err file and see if it's done. How long did it take? How would this scale if we were running it on all chromosomes?
Let's clean up the WGS data as well to keep our workspace tidy.
mkdir wgs
mv WGS* hc_wgs* wgs/
In this module we will learn about variant filtering and annotation. The raw output of GATK HaplotypeCaller will include many variants with varying degrees of quality. For various reasons we might wish to further filter these to a higher confidence set of variants. One recommended approach is to use GATK VQSR (see below for more details). This requires a large number of variants. Sufficient numbers may be available for even a single sample of whole genome sequencing data. However for targeted sequencing (e.g., exome data) it is recommended to include a larger number (i.e., at least 30-50), preferably platform-matched (i.e., similar sequencing strategy), samples with variant calls. For our purposes, we will first demonstrate a less optimal hard-filtering strategy. Then we will demonstrate VQSR filtering.
It is highly recommended to read the following documentation from GATK before applying these sorts of approaches to your own data:
First, we will separate out the SNPs and Indels from the VCF into new separate VCFs. Note that the variant type (SNP, INDEL, MIXED, etc) is not stored explicitly in the vcf but instead inferred from the genotypes. We will use a versatile GATK tool called SelectVariants. This command can be used for all kinds of simple filtering or subsetting purposes. We will run it twice to select by variant type, once for SNPs, and then again for Indels, to produce two new VCFs.
GATK SelectVariants is run with the following options:
–java-options ‘-Xmx12g’tells GATK to use 60GB of memorySelectVariantsspecifies the GATK command to run-Rspecifies the path to the reference genome-Vspecifies the path to the input vcf file to be filtered-select-type [SNP, INDEL, MIXED, etc]specifies which type of variant to limit to-Ospecifies the path to the output vcf file to be produced
cd /workspace/germline/
gatk --java-options '-Xmx12g' SelectVariants -R /workspace/inputs/references/genome/ref_genome.fa -V /workspace/germline/Exome_Norm_HC_calls.vcf -select-type SNP -O /workspace/germline/Exome_Norm_HC_calls.snps.vcf
gatk --java-options '-Xmx12g' SelectVariants -R /workspace/inputs/references/genome/ref_genome.fa -V /workspace/germline/Exome_Norm_HC_calls.vcf -select-type INDEL -O /workspace/germline/Exome_Norm_HC_calls.indels.vcfNext, we will perform so-called hard-filtering by applying a number of hard (somewhat arbitrary) cutoffs. For example, we might require each variant to have a minimum QualByDepth (QD) of 2.0. The QD value is the variant confidence (from the QUAL field) divided by the unfiltered depth of non-reference samples. With such a filter any variant with a QD value less than 2.0 would be marked as filtered in the FILTER field with a filter name of our choosing (e.g., QD_lt_2). Multiple filters can be combined arbitrarily. Each can be given its own name so that you can later determine which one or more filters a variant fails. Visit the GATK documentation on hard-filtering to learn more about the following hard filtering options. Notice that different filters and cutoffs are recommended for SNVs and Indels. This is why we first split them into separate files.
gatk --java-options '-Xmx12g' VariantFiltration -R /workspace/inputs/references/genome/ref_genome.fa -V /workspace/germline/Exome_Norm_HC_calls.snps.vcf --filter-expression "QD < 2.0" --filter-name "QD_lt_2" --filter-expression "FS > 60.0" --filter-name "FS_gt_60" --filter-expression "MQ < 40.0" --filter-name "MQ_lt_40" --filter-expression "MQRankSum < -12.5" --filter-name "MQRS_lt_n12.5" --filter-expression "ReadPosRankSum < -8.0" --filter-name "RPRS_lt_n8" --filter-expression "SOR > 3.0" --filter-name "SOR_gt_3" -O /workspace/germline/Exome_Norm_HC_calls.snps.filtered.vcf
gatk --java-options '-Xmx12g' VariantFiltration -R /workspace/inputs/references/genome/ref_genome.fa -V /workspace/germline/Exome_Norm_HC_calls.indels.vcf --filter-expression "QD < 2.0" --filter-name "QD_lt_2" --filter-expression "FS > 200.0" --filter-name "FS_gt_200" --filter-expression "ReadPosRankSum < -20.0" --filter-name "RPRS_lt_n20" --filter-expression "SOR > 10.0" --filter-name "SOR_gt_10" -O /workspace/germline/Exome_Norm_HC_calls.indels.filtered.vcfLet's take a look at a few of the variants in one of these filtered files. Remember, we haven't actually removed any variants. We have just marked them as PASS or filter-name. Use grep -v and head to skip past all the VCF header lines and view the first few records.
grep -v "##" Exome_Norm_HC_calls.snps.filtered.vcf | head -10Can you come up with a shell command that tells how many variants passed or failed our filters?
It is convenient to have all our variants in a single result file, so let's merge them back together:
GATK MergeVcfs is run with the following options:
- –java-options ‘-Xmx12g’ tells GATK to use 12GB of memory
- MergeVcfs specifies the GATK command to run
- -I specifies the path to each of the vcf files to be merged
- -O specifies the path to the output vcf file to be produced
gatk --java-options '-Xmx12g' MergeVcfs -I /workspace/germline/Exome_Norm_HC_calls.snps.filtered.vcf -I /workspace/germline/Exome_Norm_HC_calls.indels.filtered.vcf -O /workspace/germline/Exome_Norm_HC_calls.filtered.vcfIt would also be convenient to have a vcf with only passing variants. (Why don't we want this all the time?)
To create this file, we can go back the GATK SelectVariants tool. This will be run much as above except with the --exlude-filtered option instead of -select-type.
GATK SelectVariants is run with the following options:
–java-options ‘-Xmx12g’tells GATK to use 60GB of memorySelectVariantsspecifies the GATK command to run-Rspecifies the path to the reference genome-Vspecifies the path to the input vcf file to be filtered--exclude-filteredspecifies to remove all variants except those marked as PASS-Ospecifies the path to the output vcf file to be produced
gatk --java-options '-Xmx12g' SelectVariants -R /workspace/inputs/references/genome/ref_genome.fa -V /workspace/germline/Exome_Norm_HC_calls.filtered.vcf -O /workspace/germline/Exome_Norm_HC_calls.filtered.PASS.vcf --exclude-filteredHard-filtering on the WGS dataset will be performed nearly identically to above. Let's go into the wgs directory:
cd wgs
Then create a bash script called filter_wgs.sh, using vim or nano. Paste the below commands into it:
#this line provides several safeguards that instruct the script to fail if things go wrong. (if any command fails, if any variables are undefined, etc.
set -euo pipefail
# Extract the SNPs from the call set
gatk --java-options '-Xmx12g' SelectVariants -R /workspace/inputs/references/genome/ref_genome.fa -V WGS_Norm_HC_calls.vcf -select-type SNP -O WGS_Norm_HC_calls.snps.vcf
# Extract the indels from the call set
gatk --java-options '-Xmx12g' SelectVariants -R /workspace/inputs/references/genome/ref_genome.fa -V WGS_Norm_HC_calls.vcf -select-type INDEL -O WGS_Norm_HC_calls.indels.vcf
# Apply basic filters to the SNP call set
gatk --java-options '-Xmx12g' VariantFiltration -R /workspace/inputs/references/genome/ref_genome.fa -V WGS_Norm_HC_calls.snps.vcf --filter-expression "QD < 2.0" --filter-name "QD_lt_2" --filter-expression "FS > 60.0" --filter-name "FS_gt_60" --filter-expression "MQ < 40.0" --filter-name "MQ_lt_40" --filter-expression "MQRankSum < -12.5" --filter-name "MQRS_lt_n12.5" --filter-expression "ReadPosRankSum < -8.0" --filter-name "RPRS_lt_n8" -O WGS_Norm_HC_calls.snps.filtered.vcf
# Apply basic filters to the INDEL call set
gatk --java-options '-Xmx12g' VariantFiltration -R /workspace/inputs/references/genome/ref_genome.fa -V WGS_Norm_HC_calls.indels.vcf --filter-expression "QD < 2.0" --filter-name "QD_lt_2" --filter-expression "FS > 200.0" --filter-name "FS_gt_200" --filter-expression "ReadPosRankSum < -20.0" --filter-name "RPRS_lt_n20" -O WGS_Norm_HC_calls.indels.filtered.vcf
# Merge filtered SNP and INDEL vcfs back together
gatk --java-options '-Xmx12g' MergeVcfs -I WGS_Norm_HC_calls.snps.filtered.vcf -I WGS_Norm_HC_calls.indels.filtered.vcf -O WGS_Norm_HC_calls.filtered.vcf
# Extract PASS variants only
gatk --java-options '-Xmx12g' SelectVariants -R /workspace/inputs/references/genome/ref_genome.fa -V WGS_Norm_HC_calls.filtered.vcf -O WGS_Norm_HC_calls.filtered.PASS.vcf --exclude-filteredAfter loading these into the script, run it with bash filter_wgs.sh
NOTE: There are a number of MIXED type variants (multi-allelic with both SNP and INDEL alleles) that are currently dropped by the above workflow (selecting for only SNPs and INDELs). In the future we could consider converting these with gatk LeftAlignAndTrimVariants --split-multi-allelics.
A more sophisticated way to filter VCFs involves doing Variant Quality Score Recalibration (VQSR). It uses a number of reference variant call sets as truth/training data from which to build a model. This model estimates the probability that a variant is real and allows filtering at various confidence levels. This generally outperforms hard-filtering.
We will not use this technique today, but for examples of running this you can view the more comprehensive tutorial on pmbio or in the GATK documentation
Open IGV and load up your Exome Normal bam file (/workspace/align/Exome_Norm_sorted_mrkdup_bqsr.bam) and your filtered single-sample VCF file (/workspace/germline/Exome_Norm_HC_calls.filtered.PASS.vcf). Navigate to the TP53 gene, in which four variants have been called. Inspect of each of them, and answer whether the site looks like a solid call to you, and why or why not.
Some other areas of interest to look at:
chr17:7,579,249-7,579,790What's with the weird ends of reads here (make sure you have soft-clipping visible in your preferences)- What can we say about the phase of these three variants?
chr17:5,381,336-5,381,470 - How about these two?
chr17:82,870,385-82,870,452 - Or these two?
chr17:7,726,842-7,727,112
We've been using pretty heavy duty GATK commands to filter VCFs. In theory, if we wanted to do this in a more lightweight way, we could write a one-liner with grep or use a complicated regular expression to get the fields we care about, but we strongly encourage you not to do that - it's too easy to screw it up, and there are better ways!
One additional method to be aware of is bcftools, which is a powerful tool for manipulating VCF files.
As a quick example, let's just subset one of our VCFs to meet some criteria:
bcftools view --include 'MQ>55' Exome_Norm_HC_calls.filtered.PASS.vcf >high_mq.vcf
Now what if we wanted to combine that with accessing an attribute inside the INFO field?
bcftools view --include 'INFO/DP>=100 & MQ>55' Exome_Norm_HC_calls.filtered.PASS.vcf >high_mq_high_dp.vcf
How many variants did those produce compared to the original?
for file in Exome_Norm_HC_calls.filtered.PASS.vcf high_mq.vcf high_mq_high_dp.vcf;do grep -v "^#" $file | wc -l;done
These bcftools commands adhere more closely to the unix philosphy - small tools that output to the command line and can be piped to additional downstream steps. That can be convenient, especially for ad-hoc work/exploring data.
Adapted from the PMBio Germline calling exercises - see full sources and citations there.