- Introduction
- Videos
- Posts
- Papers
- De novo generators
- [Retro synthesis](#retro-synthesiPosts](#posts)
- Papers
- Software
- Data
- Labs
- Companies
- Theory
Table of contents generated with markdown-toc



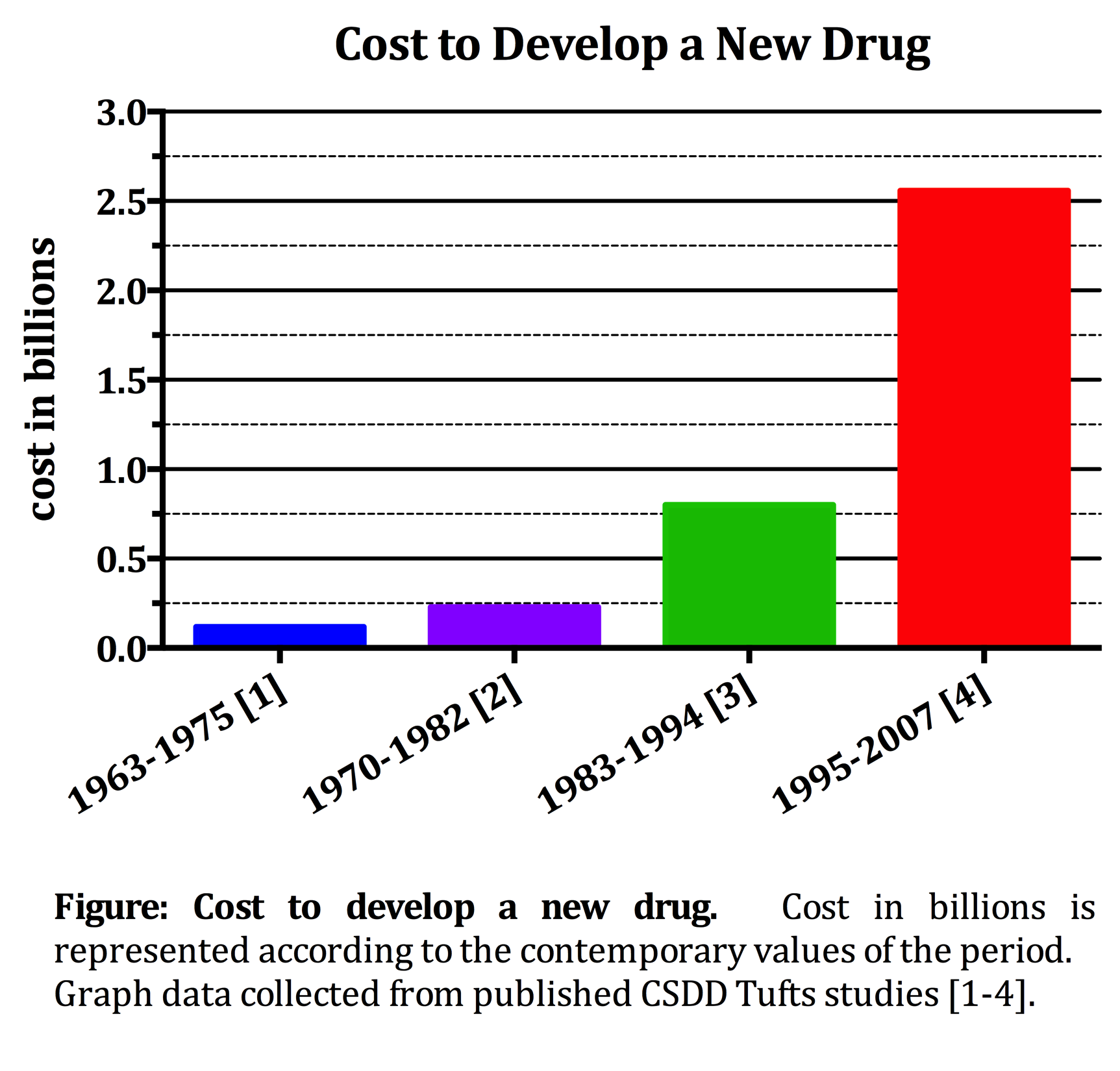

There are many startup companies
-
Make Pharma Great Again with Artificial Intelligence: some Challenges (startcrowdAI, Jun 15, 2017)
-
ChemGAN challenge for drug discovery: can AI reproduce natural chemical diversity? (startcrowdAI, Aug 30, 2017)
-
AI in drug discovery is overhyped: examples from AstraZeneca, Harvard, Stanford and Insilico Medicine (startcrowdAI, Jan 2, 2018)
-
Ratings of Labs in Artificial Intelligence for Drug Discovery (startcrowdAI, Mar 12, 2018- [ 2018)
-
MIT paper in machine learning for drug discovery at ICML 2018: very incomplete (startcrowdAI,Jul 6, 2018)
-
Automating molecule design to speed up drug development (MIT news, July 6, 2018)
DiversityNet
-
DiversityNet: a collaborative benchmark for generative AI models in chemistry (startcrowdAI, Feb 8, 2018)
-
I don’t disagree with this perspective that you’ve been putting (John Parkhill, Feb 14, 2018) "Diversity is not the issue. Data is not the issue. Physics is the issue"
-
Thank you for your detailed remarks and for sharing TensorMol (startCrowdAI, Feb 15, 2018)
-
Did you try TensorMol on the MoleculeNet benchmark? (John Parkhill, Feb 20,A new metric for generative models for molecules, by JKU Linz](https://medium.com/the-ai-lab/a-new-metric-for-generative-models-for-molecules-by-linz-university-808a73130cfc) (startcrowdAI, Apr 2 2018)
(top)
-
Deep Reinforcement Learning for de-novo Drug Design (Moscow, 2018)
Paper: Link
Notes: Data: JAK2, ChEMBL, PubChem, 14,176 (logP), 15,549 (JAK2), and 47,425 (T_m)
Method: Property prediction models, Training for the generative model, Stack-augmented recurrent neural network
Code:, Jak2 demo:
, RecurrentQSAR Jak2 Demo:
-
Prototype-Based Compound Discovery Using Deep Generative Models (Israel, 2018)
Paper: Arxiv Link, Journal link
Notes: Extend VAE to allow a conditional sampling – sampling an example from the data distribution (drug-like molecules) which is closer to a given input. Data:
Method: VAE
Code: -
Conditional Molecular Design with Deep Generative Models (Korea, 2018):
Paper: Arxiv link, Journal link
Notes:
Data: 310K Zinc, Code: -
Automatic chemical design using data driven continuous representation of molecules (Havard, 2018):
Paper: Link
Notes: , Code: -
De Novo Design at the Edge of Chaos (Schneider lab, 20??):
Paper: Link, sci-hub
Notes: , Method: Review -
Reinforced Adversarial Neural Computer for de Novo Molecular Design (Insilico, 2018):
Paper: Link
Notes: schematic view, Methods, RANC based on Organic
Code: -
Optimizing distributions over molecular space. An Objective-Reinforced Generative Adversarial Network for Inverse-design Chemistry (Organic, Havard, 2017):
Paper: Link
Notes: Method: GAN, RL Data: 15 000 drug-like form ZINC, 15 000 drug-like from ChemDiv
Code: -
Adversarial Threshold Neural Computer for Molecular de Novo Design (Insilico, 2018):
Paper: Link
Notes:
Code: -
Generating focused molecule libraries for drug discovery with RNNs (AstraZeneca, 2018):
Paper: Link
Notes:
Data: ChEMBL, Mol Representation: SMILES Methods Code: -
Exploring Deep Recurrent Models with Reinforcement Learning for Molecule Design (Benevolent.AI, 2018):
Paper: Link
Notes: , Code: -
Grammar Variational Autoencoder (Alan Turing Institute, 2017):
Paper: Link
Notes:
Code: -
Application of generative autoencoder in de novo molecular design (AstraZeneca, 2017):
Paper: Link
Notes: , Code: -
Syntax-Directed Variational Autoencoder for Structured Data (Georgia Tech, 2018):
Paper: Link
Notes:
Code: -
Deep Generative Models for Molecular Science (Georgia Tech, 2018):
Paper: Link
Notes:
Code: -
The cornucopia of meaningful leads: Applying deep adversarial autoencoders for new molecule development in oncology (Insilico, 2017):
Paper: Link
Notes:
Code: -
druGAN: an advanced generative adversarial autoencoder model for de novo generation of new molecules with desired molecular properties in silico (Insilico, 2017):
Paper: Link
Notes:
Code: -
De novo drug design with deep generative models : an empirical study (??, 2017):
Paper: Link
Notes:
Code: -
De novo drug design with deep generative models : an empirical study (??, 2017):
Paper: Link
Notes: RNN generative models for stochastic optimization in the context of de novo drug design.
Code: -
Molecular generation with recurrent neural networks (Wildcard consulting, 2017):
Paper: Link
Notes: RNN with LSTM cells can generate synthesizable molecules.
Code: -
Sequence Tutor: Conservative Fine-Tuning of Sequence Generation Models with KL-control (Google, 2017):
Paper: Link
Notes: Method for improving sequence generated by a RNN
Code: -
Molecular De Novo Design through Deep Reinforcement Learning (AstraZeneca, 2017):
Paper: Link
Notes: Sequence-based generative model for molecular de novo design.
Code: -
ChemTS: An Efficient Python Library for de novo Molecular Generation (Tokyo, 2017):
Paper: Link
Notes: Python library ChemTS that explores the chemical space by combining Monte Carlo tree search (MCTS) and an RNN.
Code: -
Generative Recurrent Networks for De Novo Drug Design (Schneider lab, 2017):
Paper: Link
Notes: De novo design that utilizes RNN containing LSTM cells.
Code: -
Molecular generative model based on conditional variational autoencoder for de novo molecular design (KAIST Korea, 2018):
Paper: Link
Notes: Conditional variational autoencoder CVAE) for de novo molecular design (5 properties,Aspirin, Tamiflu).
Code: -
Improving Chemical Autoencoder Latent Space and Molecular De novo Generation Diversity with Heteroencoders (Wildcard consulting, 2018):
Paper: Link
Notes: Dataset: GDB-8 dataset.
Code:


-
Towards "AlphaChem": Chemical Synthesis Planning with Tree Search and Deep Neural Network Policies:
Paper: link -
Learning to Plan Chemical Syntheses:
Paper: link -
Retrosynthetic Reaction Prediction Using Neural Sequence-to-Sequence Models:
Paper: link, Code: link -
Planning chemical syntheses with deep neural networks and symbolic AI:
Paper: link -
Predicting Organic Reaction Outcomes with Weisfeiler-Lehman Network
Paper: link, Code: link
| Paper | Author | Year | Abstract |
|---|---|---|---|
| De Novo Design at the Edge of Chaos | Schneider lab | Current perspective automated molecule generation. | |
| Reinforced Adversarial Neural Computer for de Novo Molecular Design | Insilico | 2018 | Reinforced Adversarial Neural Computer (RANC) |
| Optimizing distributions over molecular space. An Objective-Reinforced Generative Adversarial Network for Inverse-design Chemistry | Havard | 2017 | Objective-Reinforced Generative Adversarial Networks (ORGANIC) |
| Adversarial Threshold Neural Computer for Molecular de Novo Design | Insilico | 2018 | Adversarial Threshold Neural Computer (ATNC), de novo design of novel small-molecules (Generative Adversarial Networks (GANs) with Reinforcement Learning) |
| Generating focused molecule libraries for drug discovery with RNNs | AstraZeneca | 2018 | RNNs can be trained as generative models for molecular structures |
| Exploring Deep Recurrent Models with Reinforcement Learning for Molecule Design | Benevolent.AI | 2018 | 19 benchmarks, apply reinforcement learning techniques for molecular design |
| Automatic chemical design using data driven continuous representation of molecules | Havard | 2018 | Convert molecules to and from a multidimensional continuous representation. |
| Conditional Molecular Design with Deep Generative Models | 2018 | Conditional molecular design method that facilitates generating new molecules with desired properties. | |
| Grammar Variational Autoencoder | Alan Turing Institute | 2017 | VAE using parse trees to check validity. |
| Application of generative autoencoder in de novo molecular design | AstraZeneca | 2017 | Performance of various autoencoders as generators |
| Syntax-Directed Variational Autoencoder for Structured Data | Georgia Tech | 2018 | Syntax-directed variational autoencoder (SD-VAE) with on-the-fly generated guidance for constraining the decoder |
| Deep Generative Models for Molecular Science | Denmark Tech | 2018 | Review deep generative models for predicting molecular properties. Focus on autoencoder |
| The cornucopia of meaningful leads: Applying deep adversarial autoencoders for new molecule development in oncology | Insilico | 2017 | First application of AAE for generating novel molecular fingerprints with a defined set of parameters. |
| druGAN: an advanced generative adversarial autoencoder model for de novo generation of new molecules with desired molecular properties in silico | Insilico | 2017 | AAE and its advantages compared to VAE |
| De novo drug design with deep generative models : an empirical study | 2017 | RNN generative models for stochastic optimization in the context of de novo drug design. | |
| Molecular generation with recurrent neural networks | Wildcard consulting | 2017 | RNN with LSTM cells can generate synthesizable molecules. |
| Sequence Tutor: Conservative Fine-Tuning of Sequence Generation Models with KL-control | 2017 | Method for improving sequence generated by a RNN | |
| Molecular De Novo Design through Deep Reinforcement Learning | AstraZeneca | 2017 | Sequence-based generative model for molecular de novo design. |
| ChemTS: An Efficient Python Library for de novo Molecular Generation | Tokyo | 2017 | Python library ChemTS that explores the chemical space by combining Monte Carlo tree search (MCTS) and an RNN |
| Generative Recurrent Networks for De Novo Drug Design | Schneider lab | 2017 | De novo design that utilizes RNN containing LSTM cells. |
| Molecular generative model based on conditional variational autoencoder for de novo molecular design | KAIST Korea | 07.2018 | conditional variational autoencoder CVAE) for de novo molecular design (5 properties,Aspirin, Tamiflu). |
- De Novo Design of Bioactive Small Molecules by Artificial Intelligence
- Exploring Deep Recurrent Models with Reinforcement Learning for Molecule Design (Benevolent.AI), comment
It’s good to start with models already tried in the literature:
- Variational auto-encoder: Harvard 1, Alan Turing Institute, AstraZeneca 3, Georgia Tech, Denmark Tech (use Sci-Hub for the paywall)
- Adversarial auto-encoder: In SilicoMedicine 1, InsilicoMedicine 2 (DruGAN) (use Sci-Hub to bypass the paywall), AstraZeneca 3
- Recurrent Neural Networks (RNN): Paris-Saclay, Wildcard
- Reinforcement Learning (RL)+ RNN: Google, AstraZeneca 1, AstraZeneca 2, University of Tokyo, ETH Zurich 1, University of North Carolina, Novartis, ETH Zurich 2
- RL+ RNN+ Generative Adversarial Networks (GAN): Harvard 2 (ORGAN), Harvard 3 (ORGANIC)
- Conditional Graphs: Peking University
For GAN, there are different flavors: Wasserstein-GAN (Facebook), Cramer-GAN (DeepMind), Optimal Transport-GAN (OpenAI), Coulomb-GAN (Linz University), although at the end, maybe they are all equal (Google).
(top)
You can also find more in the Natural Language Processing literature (and apply them to SMILES):
- Texygen benchmark (Shanghai University)
- MaskGAN (Google)
- ACtuAL (University of Montreal)
- ARAE (New York University)
- Adversarial Generation of Natural Language (University of Montreal) (and don’t miss the adversarial review)
- MaliGAN (University of Montreal)
- RankGAN (University of Washington)
- GSGAN (Alan Turing Institute)
- TextGAN (Duke University)
- LeakGAN (Shanghai University)
-
Benevolent AI drug discovery paper at ICLR 2018: my open review
-
ChemGAN challenge for drug discovery: can AI reproduce natural chemical diversity?
(top)
molecule generator (wildcard consulting) (top)
In most papers, data is taken from:
-
Automatic Chemical Design Using a Data-Driven Continuous Representation of Molecules
-
Learning Graph-Level Representation for Drug Discovery (paper)
-
Molecular generative model based on conditional variational autoencoder for de novo molecular design
-
MolGAN: An implicit generative model for small molecular graphs
-
Syntax-Directed Variational Autoencoder for Structured Data, https://arxiv.org/abs/1802.08786, openreview: link (top)
Rating of labs in AI for drug discovery
(top)
101 Startups Using Artificial Intelligence in Drug Discovery
Generate Novel Drug Candidates
From a computational perspective, druglike molecular structure can be represented in five ways.
- SMILES(41) or InChi,(42)
- molecular fingerprint,(43)
- set of molecular descriptors, such as molecular weight, logP, number of heavy atoms, number of rotatable bonds, etc.,
- graph in which atoms are nodes and links are bonds between atoms, or
- 3D electron density map
Comparison
aka simplified molecular-input line-entry systems

{kind=link}
{kind=link}
- One fingerprint can match several molecules, so there is no one-to-one mapping from a molecule to the fingerprint,
- The fingerprint representation contains less information about the molecule topology than the string representation
(top)
Designing evaluation metrics is an important part of the challenge. These metrics assess the quality and diversity of generated samples. Here, contributions from medicinal chemists and statisticians are especially welcome.
Measures of diversity are based on distance metrics in the chemical space. This distance tells when two molecules are chemically close to each other. The most popular distance is the Tanimoto distance on Morgan fingerprints. It’s not necessary to get into details of the definition, the point is that those fingerprints are hand-crafted features, and it’s probably better to replace them with deep learning features, as suggested in the MoleculeNet benchmark.
Let’s denote:
- Td the distance in the chemical space.
- A the set of generated molecules with desired properties. Its size is noted |A|.
- B the training set.
it’s the average distance between a generated molecule in A and its nearest neighbor in the training set B. The formula is:
NN(A,B)=\frac{1}{|A|}\sum_{x\in A}\min_{y\in B}T_{d}(x,y)NN(A,B)=∣A∣1x∈A∑y∈BminTd(x,y)
it’s the average distance of desired generated molecules with each other. The formula is:
I(A)=\frac{1}{|A|^{2}}\sum_{(x,y)\in A\times A}T_{d}(x,y)I(A)=∣A∣21(x,y)∈A×A∑Td(x,y)
Another measure of internal diversity is to compare the set of generated samples with a reference set, which is known to be diverse a priori. For example, the ZINC dataset seems suitable. Chemists can propose alternative reference datasets.
The idea is to take a random subset of the reference set with the same size as the generated set. Then to consider those two sets as two piles of sand in the chemical space, and measure the energy necessary to move the first pile into the second pile (this measure is known as Earth Mover Distance in statistics, and Wasserstein metric in mathematics).
OpenAI. This metric uses the Inception predictive model, which is a standard image classifier (a winner of the ImageNet challenge). A generative model has a high Inception score when the Inception model is very confident that generated images belong to a particular ImageNet category, and when all categories are equally represented. This suggests that the generative model has both high quality and diversity.
(Linz University): it computes a distance between distributions of the training data and of the generated data. See their Fréchet ChEMBLNet distance.
(top)
Modeling Molecules with Recurrent Neural Networks
Learns a distribution normal bottleneck vector z is replaced by two vectors
- mean
- standard deviation
Loss function: Reconstruction loss KL divergence (makes sure distribution you're learning isn't too far from normal distribution)
Disentangled autoencoder
Github code: Molecule generator chembl autoencoder
Paper to reimplement: paper, code
Starting points: MNIST: Pytorch VAE example, another, more detailled Convolutional autoencoder (exercise/solution) Denoising autoencoder (exercise/solution)
pytorch/VAE Graph decoders DeepChem issue, icml18-jtnn, github 2
Theory VAE explained
(top)
Video: here
http://iktos.ai/successful-lead-optimization-project-in-collaboration-with-servier-presented-at-efmc2018/ Video: here Poster: here
| A | B | C |
|---|---|---|
 |
 |
 |
 |
 |
- Goal: Associate defined structural modifications with chemical property changes, including biological activity (SAR)
- It is argued that longer matched series is more likely to exhibit preferred molecular transformation while, matched pairs exhibit only a small preference

|
-
large change in potency that correspond to small changes in the molecular structures
-
high SAR information content
Literature McPairs software youtube
 |
 |
 |
 |
(top)
Putin, E., et al. (2018). "Reinforced Adversarial Neural Computer for de Novo Molecular Design." Journal of chemical information and modeling.
 |
 |
 |
 |
 |
 |
(top)
Benjamin, S.-L., et al. (2017). Optimizing distributions over molecular space. An Objective-Reinforced Generative Adversarial Network for Inverse-design Chemistry (ORGANIC).
 |
Putin, E., et al. (2018). "Adversarial Threshold Neural Computer for Molecular de Novo Design." Molecular pharmaceutics.
 |
 |
 |
 |
| Idea: adsasadsad |
Methods |
|
Results |
|
|
|
Generating focused molecule libraries for drug discovery with recurrent neural networks.
| In _de novo_ drug design, computational strategies are used to generate novel molecules with good affinity to the desired biological target. In this work, we show that recurrent neural networks can be trained as generative models for molecular structures, similar to statistical language models in natural language processing. We demonstrate that the properties of the generated molecules correlate very well with the properties of the molecules used to train the model. In order to enrich libraries with molecules active toward a given biological target, we propose to fine-tune the model with small sets of molecules, which are known to be active against that target. Against _Staphylococcus aureus_, the model reproduced 14% of 6051 hold-out test molecules that medicinal chemists designed, whereas against _Plasmodium falciparum_ (Malaria), it reproduced 28% of 1240 test molecules. When coupled with a scoring function, our model can perform the complete _de novo_ drug design cycle to generate large sets of novel molecules for drug discovery. |  |
 |
 |
 |
(top)
Neil, D., et al. (2017). "EXPLORING DEEP RECURRENT MODELS WITH REINFORCEMENT."
 |
 |
 |
Gomez-Bombarelli, R. "Automatic chemical design using data driven continuous representation of molecules."
 |
 |
 |
 |
 |
 |
(top)
Kang, S. and K. Cho (2018). "Conditional Molecular Design with Deep Generative Models." J Chem Inf Model.
 |
 |
 |
 |
 |
 |
Matt J. Kusner, et al. (2017). "Grammar Variational Autoencoder."
 |
 |
 |
(top)
Blaschke, T., et al. (2017). "Application of generative autoencoder in de novo molecular design."
 |
 |
 |
 |
 |
 |
(top)
Dai, H., et al. (2018). "Syntax-Directed Variational Autoencoder for Structured Data."
 |
 |
 |
 |
 |
 |
 |
 |
(top)
Jorgensen, P. B., et al. (2018). "Deep Generative Models for Molecular Science." Mol Inform 37(1-2).
We review these recent advances within deep generative models for predicting molecular properties, with particular focus on models based on the probabilistic autoencoder (or varia- tional autoencoder, VAE) approach in which the molecular structure is embedded in a latent vector space from which its properties can be predicted and its structure can be restored.
(top)
Kadurin, A., et al. (2017). "The cornucopia of meaningful leads: Applying deep adversarial autoencoders for new molecule development in oncology." Oncotarget
 |
 |
Kadurin, A., et al. (2017). "druGAN: an advanced generative adversarial autoencoder model for de novo generation of new molecules with desired molecular properties in silico." Molecular pharmaceutics 14(9): 3098-3104.
 |
 |
 |
 |
De novo drug design with deep generative models : an empirical study
Bjerrum, E. J. and R. Threlfall (2017). "Molecular generation with recurrent neural networks."
 |
 |
 |
(top)
Sequence Tutor: Conservative Fine-Tuning of Sequence Generation Models with KL-control
| This paper proposes a general method for im- proving the structure and quality of sequences generated by a recurrent neural network (RNN), while maintaining information originally learned from data, as well as sample diversity. An RNN is first pre-trained on data using maximum likeli- hood estimation (MLE), and the probability dis- tribution over the next token in the sequence learned by this model is treated as a prior pol- icy. AnotherRNNis then trained using reinforce- ment learning (RL) to generate higher-quality outputs that account for domain-specific incen- tives while retaining proximity to the prior pol- icy of the MLE RNN. To formalize this objec- tive, we derive novel off-policy RL methods for RNNs from KL-control. The effectiveness of the approach is demonstrated on two applications; 1) generating novel musical melodies, and 2) com- putational molecular generation. For both prob- lems, we show that the proposed method im- proves the desired properties and structure of the generated sequences, while maintaining informa- tion learned from data. |  |
 |
(top)
| This paper proposes a general method for im- proving the structure and quality of sequences generated by a recurrent neural network (RNN), while maintaining information originally learned from data, as well as sample diversity. An RNN is first pre-trained on data using maximum likeli- hood estimation (MLE), and the probability dis- tribution over the next token in the sequence learned by this model is treated as a prior pol- icy. AnotherRNNis then trained using reinforce- ment learning (RL) to generate higher-quality outputs that account for domain-specific incen- tives while retaining proximity to the prior pol- icy of the MLE RNN. To formalize this objec- tive, we derive novel off-policy RL methods for RNNs from KL-control. The effectiveness of the approach is demonstrated on two applications; 1) generating novel musical melodies, and 2) com- putational molecular generation. For both prob- lems, we show that the proposed method im- proves the desired properties and structure of the generated sequences, while maintaining informa- tion learned from data. |  |
 |
 |
 |
 |
 |
(top)
ChemTS: An Efficient Python Library for de novo Molecular Generation
| Automatic design of organic materials requires black-box optimization in a vast chemical space. In conventional molecular design algorithms, a molecule is built as a combination of predetermined fragments. Recently, deep neural network models such as variational auto encoders (VAEs) and recurrent neural networks (RNNs) are shown to be effective in de novo design of molecules without any predetermined fragments. This paper presents a novel python library ChemTS that explores the chemical space by combining Monte Carlo tree search (MCTS) and an RNN. In a benchmarking problem of optimizing the octanol-water partition coefficient and synthesizability, our algorithm showed superior efficiency in finding high-scoring molecules. ChemTS is available at https://github.com/tsudalab/ChemTS. |  |
 |
(top)
Generative Recurrent Networks for De Novo Drug Design
| Generative artificial intelligence models present a fresh approach to chemogenomics and _de novo_ drug design, as they provide researchers with the ability to narrow down their search of the chemical space and focus on regions of interest. We present a method for molecular _de novo_ design that utilizes generative recurrent neural networks (RNN) containing long short‐term memory (LSTM) cells. This computational model captured the syntax of molecular representation in terms of SMILES strings with close to perfect accuracy. The learned pattern probabilities can be used for _de novo_ SMILES generation. This molecular design concept eliminates the need for virtual compound library enumeration. By employing transfer learning, we fine‐tuned the RNN′s predictions for specific molecular targets. This approach enables virtual compound design without requiring secondary or external activity prediction, which could introduce error or unwanted bias. The results obtained advocate this generative RNN‐LSTM system for high‐impact use cases, such as low‐data drug discovery, fragment based molecular design, and hit‐to‐lead optimization for diverse drug targets. |  |
 |
 |
 |
- Goal: Associate defined structural modifications with chemical property changes, including biological activity (SAR)
- It is argued that longer matched series is more likely to exhibit preferred molecular transformation while, matched pairs exhibit only a small preference
|
|
|
-
large change in potency that correspond to small changes in the molecular structures
-
high SAR information content
Literature McPairs software youtube
Written with StackEdit.